化學所在惰性碳氫鍵活化研究中取得系列進展
碳氫鍵是一類基本的化學鍵,存在于幾乎所有的有機化合物中。碳氫鍵的鍵能非常高,碳元素與氫元素的電負性又很接近,因而碳氫鍵的極性很小,這些因素使得碳氫鍵具有惰性,在溫和條件下將碳氫鍵選擇性催化活化、構建其它含碳化學鍵存在熱力學和動力學的雙重挑戰,是化學研究的一個基本問題,也是制約分子合成和制備獲得重大突破的瓶頸問題。為了深入研究控制碳氫鍵活化轉化的物理化學本質,進而理性設計催化劑,實現高效、綠色的碳氫鍵活化轉化,中國科學院化學研究所在物理化學、計算化學、有機化學領域布置了研究力量并取得了系列研究進展。
1. 研制原子團簇實驗儀器并成功應用于甲烷碳氫鍵活化研究
甲烷是天然氣的主要成分,甲烷的活化轉化具有重大的應用需求,然而甲烷分子具有四面體對稱性,其碳氫鍵特別穩定,因而甲烷在溫和條件下的活化轉化是一個科學難題。化學所研究員何圣貴及其合作者從頭設計并建立了原子團簇制備、化學反應和結構表征實驗系統,在可控、可重復、排除外界不確定因素干擾的條件下,測定一系列原子團簇與甲烷的反應活性,結合理論化學計算,探索發現能夠活化轉化甲烷的關鍵幾何和電子結構及其性能調控因素。研究發現金屬團簇上的原子氧自由基可以在室溫條件下有效活化甲烷并產生甲基自由基,該反應的活性受到氧自由基局部電荷和自旋的控制(Acc. Chem. Res. 2012, 45, 382-390; Angew. Chem. Int. Ed.2013, 52, 2444-2448);氧化鋁、氧化鈦等團簇上的鉑原子或金原子,可以有效活化甲烷,進而在載體團簇的共同作用下生成甲醛分子(Angew. Chem. Int. Ed. 2014, 53, 9482-9486; Chem. Sci.2016, 7, 4730-4735; J. Am. Chem. Soc. 2016, 138, 9437-9443)。
金屬中心的氧化加成反應是C-H鍵活化的重要方式之一,這類反應常常發生在具有配對價電子的后過渡金屬體系,特別是貴金屬體系,而前過渡金屬中心價電子數目相對少、難配對,因而前過渡金屬體系缺乏甚至完全沒有氧化加成反應性。科研人員使用團簇質譜、團簇光電子速度成像譜,結合合作者陳輝等的高精度理論計算研究,發現通過選擇合適的配體,調控前過渡金屬Nb和Mo的電子結構,能夠顯著降低金屬中心具有配對電子的低自旋態能級,直接或間接參與碳氫鍵的氧化加成反應(圖1),實現甲烷分子在室溫條件下的活化轉化(Angew. Chem. Int. Ed. 2016, 55, 5760-5764;Angew. Chem. Int. Ed. 2016, 55, 4947-4951),該研究工作為非貴金屬替代貴金屬活化碳氫鍵提供了理論依據。
2. 構建碳氫鍵鐵基催化活化新理論和反應新體系
鐵是地球乃至宇宙中含量最豐富的過渡金屬,具有廉價易得、環境友好等優點,鐵基催化廣泛存在于生物體和工業生產中。在金屬有機化學領域,鐵基催化特別是低價鐵催化活化碳氫鍵受到廣泛關注,但對其反應機理和反應性本質的認識還非常缺乏,這是由于鐵中心體系常具有復雜的開殼層電子結構以及多個能量相近的自旋態,是理論和計算化學研究的難點。針對低價Fe催化活化C(sp2)-H和C(sp3)-H鍵的芳基化反應,利用密度泛函與耦合簇方法相結合的計算化學策略,化學所研究員陳輝及其合作者提出了二態反應性圖像(圖2)。研究發現:(1)二價Fe和三價Fe都能以s-鍵復分解的模式,通過在反應物端的低自旋激發態介導碳氫鍵的活化;(2)在碳氫鍵活化后,反應中的二鹵代烴氧化劑通過單電子轉移(SET)的機理將二價Fe氧化為三價Fe,三價Fe的形成對后續C-C偶聯過程的順利發生必不可少;(3)在三價Fe促發的還原消除C-C偶聯過程之后,反應中的二鹵代烴氧化劑繼續通過SET機理將生成的一價Fe氧化再生為二價或三價Fe,與SET相比,雙電子參與的氧化加成過程在能量上是不利的;(4)整個催化循環中,Fe的氧化態演化過程為Fe(II)/Fe(III)/Fe(I)或Fe(III)/Fe(I),配體具有穩定高活性的低自旋態Fe物種的作用(J. Am. Chem. Soc. 2016, 138, 3715-3730)。
針對低價的多核鐵催化的碳氫活化反應,化學所研究員王從洋、陳輝及其合作者通過合成化學實驗和計算化學理論模擬相結合,實現了羰基鐵催化的N-H亞胺與炔烴的氧化還原中性[4+2]環化反應,高效生成順式3,4-二氫異喹啉化合物。該反應不需要任何額外添加的堿、配體或其他添加物,體現了完美的原子經濟性。催化機理研究表明,羰基鐵-鐵間的雙核協同催化是實現該反應的關鍵因素,為多核金屬催化碳氫鍵活化開辟了新的方向(Angew. Chem. Int. Ed. 2016, 55, 5268-5271)。
3. 碳氫鍵錳族金屬催化活化反應新體系
金屬錳具有來源豐富、價格便宜等優點,但目前錳的金屬有機化學研究主要局限于當量反應。如何提高有機錳化合物的反應活性,控制其反應的選擇性,特別是構建完整的錳催化循環是該領域研究的挑戰性難題。化學所研究員王從洋及其合作者采用有機分子與錳族金屬協同及雙金屬協同等策略,發展了一系列錳族金屬催化的C-H鍵活化轉化新反應,展示了其不同于其他過渡金屬的獨特反應性(圖3):(1)采用錳催化劑和有機分子—二環己基胺的組合,發展了錳/胺協同脫質子的C-H活化新模式;通過錳配體間質子轉移及分子內炔基輔助錳實現C-H活化等關鍵步驟,構建了完整的錳催化循環;同時,用計算化學方法驗證了此催化循環的合理性(J. Am. Chem. Soc. 2013, 134, 1264-1267);(2)使用錳族金屬與主族金屬的雙金屬連續催化策略,實現了苯甲酰胺和炔烴的中性[4+2]環化反應;通過反應條件的調控,可以高非對映選擇性分別得到順-3,4-二氫異喹啉酮和反-3,4-二氫異喹啉酮化合物,體現了該催化體系高度的靈活性(J. Am. Chem. Soc. 2013, 135, 4628-4631);(3)發展了錳催化的N-H亞胺與炔烴的[4+2]環化反應高效生成異喹啉化合物;與已有過渡金屬催化的C-H活化合成異喹啉方法不同,該反應的副產物是氫氣,具有很高的原子經濟性(Angew. Chem. Int. Ed. 2014, 53, 4950-4953);(4)利用錳/鎂雙金屬體系將四氫呋喃形式上轉化為1-丁醇-4-卡賓的等量體,其同時與親電試劑(亞胺/腈)和親核試劑(格氏試劑)反應,構建了同碳上兩個新的C-C鍵,從而高效制備了1,5-氨基醇或1,5-酮醇化合物(J. Am. Chem. Soc. 2014, 136, 6558-6561);(5)通過錳催化劑與路易斯酸的雙重活化策略,實現了惰性C(sp2)−H鍵對醛和腈的“格氏型”親核加成反應;該反應條件溫和,具有很好的區域選擇性和立體選擇性,同時底物適用范圍廣泛,具有良好的官能團容忍性(Angew. Chem. Int. Ed. 2015, 54, 13659-13663)。
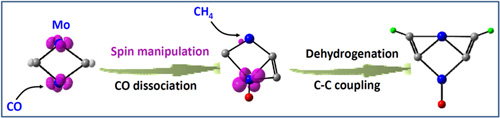
圖1 一氧化碳解離吸附調控Mo金屬中心的自旋態,實現甲烷的活化轉化
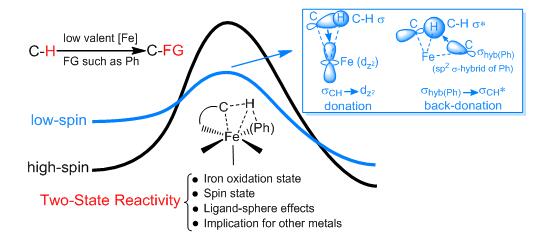
圖2 低價鐵催化活化碳氫鍵的二態反應性圖像
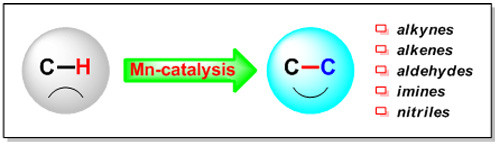
圖3 新型錳族金屬催化體系的建立及其活化碳氫鍵的獨特反應性
標簽:
相關資訊
2、如涉及作品內容、版權和其它問題,請在30日內與本網聯系,我們將在第一時間作出適當處理!有關作品版權事宜請聯系:+86-571-88970062