大連化物所金屬表面解離吸附動力學理論研究取得新進展
近日,中國科學院大連化學物理研究所分子反應動力學國家重點實驗室在分子表面散射動力學理論研究中獲得新進展。由該實驗室副研究員傅碧娜、研究員張東輝等撰寫的論文First-principles quantum dynamical theory for the dissociative chemisorption of H2O on rigid Cu(111) 發表在近期的《自然·通訊》雜志上(Nature Communications, 2016, 7:11953, doi: 10.1038/ncomms11953),該研究工作首次實現了多原子分子在金屬表面反應的全維量子動力學計算。
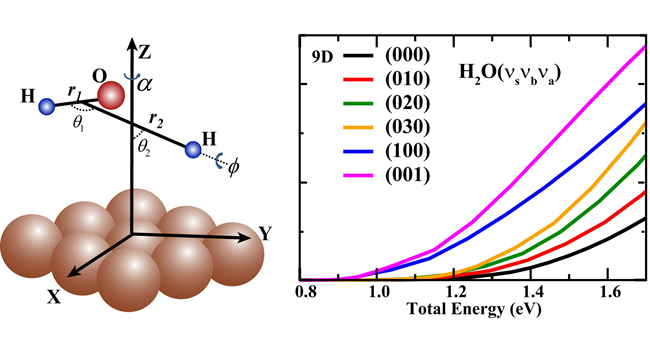
分子在金屬表面解離吸附的動力學研究在多相催化等工業過程中占有重要的地位。在過去的20多年里,科學家們為發展可靠的理論來精確描述分子在固體表面的解離吸附動力學付出了巨大的努力。由于反應中可能存在的量子效應,如量子隧穿、零點能、反應共振等,量子動力學研究是最為可靠的。但是由于高維量子動力學研究的困難,以往精確的量子動力學理論只局限于研究雙原子分子在固體表面解離吸附這類包含六個自由度的問題。水在過渡態金屬表面的解離吸附是多相催化過程,如水煤氣變化和蒸氣重整反應中重要的一步,因此其研究具有重要意義。由于包含9個自由度,以往的研究只能利用減維量子模型把體系的自由度限制在六個。今年初,該研究團隊首次利用7維量子動力學方法研究H2O在Cu(111)表面的解離吸附動力學 (Chemical Science, 2016, 7, 1840-1845),發現7維量子動力學結果和之前的6維結果相差很大,說明表面減維模型在描述此類反應時會帶來較大的誤差,因此非常有必要開展全維量子動力學研究。
最近,該研究團隊在其擬合的全維全域勢能面上成功在全維(9維)水平計算了H2O在Cu(111)表面的解離吸附幾率,從而首次實現了一個三原子分子在金屬表面反應的全維量子動力學研究,被審稿人譽為“理解表面反應動力學的一個重要里程碑”。他們的研究發現全維量子解離幾率與以往用減維模型得到的結果相差很大,證明只有全維量子計算才能精確描述此類反應。全維量子動力學還揭示了H2O的不同振動模式激發比平動能都能更有效地促進反應發生,并且效率明顯要比減維模型得到的更為顯著。該全維量子動力學研究也驗證了該研究團隊之前在雙原子分子-表面散射中所發展的質心位點平均方法的準確性,表明該方法能廣泛應用到多原子分子在金屬表面的解離吸附動力學研究中,從而為精確模擬多原子分子-表面反應提供了一個重要的理論方法。
以上研究得到了國家自然科學基金委、科技部和中科院的支持。
大連化物所金屬表面解離吸附動力學理論研究取得新進展
標簽:
相關資訊
2、如涉及作品內容、版權和其它問題,請在30日內與本網聯系,我們將在第一時間作出適當處理!有關作品版權事宜請聯系:+86-571-88970062